摘要由于DNA序列双螺旋结构的互补对称性质以及空间上的复杂结构,探索长距离DNA片段的匹配特征尤其是在互补关系上有着重要的意义。本文利用子串–互补串匹配技术通过对DNA序列已有结构的检测分析着重对回文序列、发夹结构等可能存在的结构进行预测。通过统计测量的方法对DNA数据进行处理,对结果进行对比分析,将测量数据转换为可视图,对批量复杂DNA序列的提取特征数据进行可视化分析。通过结果图示,可以看到选择的DNA序列中的确存在着长距离匹配结构。文中给出的可视化方法,提出的分析测量模型以及提取的测量特征可视化机制能为后续不同DNA序列数据以及结构的可视化分析的应用研究提供坚实的模型和实践基础。
关键词DNA空间结构,长距离匹配,子串–互补串匹配,可视化
1.引言
自1980年的噬菌体Φ-X174实现完全测序,成为第一个测定的基因组以来,伴随着基因组学以及测序技术的发展,尤其是人类基因组计划从20世纪90年代启动到2000年人类基因组草图正式完成,再至2005年人类基因组计划的测序工作基本完成,已经有了难以计数的生物信息尚待人们挖掘与探索。面对海量的数据,在分子生物学的基础上,生物信息学[1]应运而生。生物信息学综合了各种理论和技术,主要对生物信息进行采集,处理,传播,分析和解释。而研究DNA序列的结构以及由A、T、C、G组成的碱基序列中的特征和规律是生物信息学中相当重要的课题。
在目前对于DNA序列的特征提取分析以及相似性比较的研究中,诸多有效的方法和工具正在不断涌现。如骆家伟等学者通过对DNA序列计算信息离散度来进行相似性分析[2]。同时利用可视化工具对DNA序列进行分析也是目前的一大热点。如白兰凤提出的利用二维图表示DNA序列,计算对应的距离矩阵并得到特征值以比较特征[3]。还有郑智捷等一批学者使用流密码的随机性检测方法以及变值逻辑体系对DNA序列进行可视化分析,并取得了一系列成果[4]-[7]。而随着目前计算机技术的发展与成熟诸如神经网络等技术也被应用在了如DNA序列分类中[8]。
而在本文中则区别于其他方法,将重点集中在DNA序列的对称和互补现象中,采用了子串–互补串匹配技术,针对特有的DNA序列结构如回文结构发夹结构,对DNA序列中的长距离匹配现象进行检索和分析,并通过可视化等一系列方式挖掘其中隐藏的信息,直接从序列一级结构层次上对可能出现的空间结构特征进行探索,为DNA序列精细特征的分析研究提供了一种新的方法。
2.共轭对称及回文结构
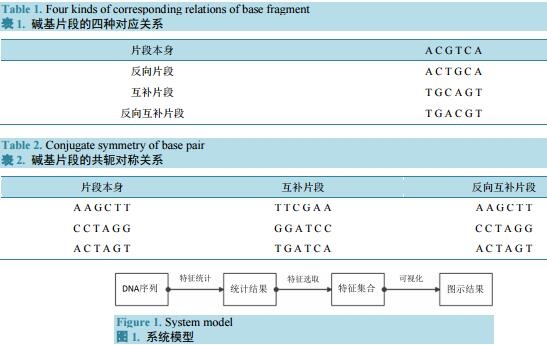
在DNA序列中,碱基配对遵从着严格的互补对称关系,本文所研究的重点主要在DNA碱基序列上,为了找出具有特殊性的序列上的碱基片段,着重研究四种片段形式,分别是:片段本身,反向片段,互补片段,反向互补片段。以片段ACGTCA为例,其四种形式如表1所示。
此四种形式的片段并非本文中第一次提出,同时也存在其它形式的片段,但在本文研究中,为了研究因互补配对造成的可能存在的空间结构,故而选取这种较基本的片段形式进行后续的搜索统计和观察。
同时,需要注意的是在诸多片段中,存在着一种较为特殊的片段,具体表现在该片段的反向互补片段是它的本身。如表2所示。
若出现表2情况的片段,则称其为片段与反向互补片段之间存在共轭对称关系。
而回文结构在生物基因组学中主要是指双链DNA中某段序列具有的反向重复的结构,当该序列的双链被打开后,可形成发夹结构。这段序列则被称为回文序列(Palindromicsequence)。
其特点是在该段的碱基序列的互补链之间正向与反向是相同的,
例如5’AAGCTT’3
3’TTCGAA’5
在双链中,可能如下方序列所示出现:
……CGATTACAGGCTAAGCTTTCCAGCGTACACG……
……GCTAATGTCCGATTCGAAAGGTCGCATGTGC……
3.系统和方法
本文建立的系统包括3个核心模块:特征统计模块、特征分析模块和可视化模块。处理过程为把若干段等长的序列分别进行特征统计,再从统计结果中选出具有代表性的特征,最后对选取得特征集合进行可视化,显示各序列中特征片段的分布特征。该系统的结果和处理过程如图1所示。
3.1.数据处理模块
在诸多对DNA数据分析测量的研究中,使用了诸如数据挖掘,基于统计,基于概率等的各种方法来进行探索,而在对基因回文结构的研究探索中,期待能够发现回文结构并对其位置以及序列形式进行记录和统计分析,以得出此类结构是否存在某种特殊现象及分布规律,故而选择基于统计的方式来探索其特性。通过对对应序列之间的匹配,寻找两条序列之间是否存在由同种类型的碱基片段形成的回文结构,并观察其在同一数据集中的特征。最后用统一的数据结构保存得到的信息,为统计出现在数据集中的位置分布情况以及出现次数以及可视化提供数据。
本文中的匹配模式,即以数据集中的单条或成对DNA序列作为处理对象,在序列中通过滑动窗口的形式依次选择碱基片段,对于序列中选取碱基片段位置之后出现的对应形式碱基片段记录位置,从而实现匹配处理,得到关于数据集中每个出现碱基片段的所有形式的位置数据。匹配过程如图2所示。
模块的输入:
以FASTA格式保存记录的两组若干等数量等长度DNA序列集合,两组序列皆取自某基因左右两端各500bp长DNA序列。每条编号的序列皆有另一条编号相同的序列与其对应。
模块的匹配处理:
从两组数据集中依次选取左右两条对应编号的DNA序列,使左右两条序列各自对自身序列进行匹配,记录片段位置信息,同时将左侧序列与右侧序列进行匹配,初始匹配片段长度从3开始,直至出现长度为n的碱基片段使得该序列中不存在有任意长度为n+1的片段在对应序列中存在与其匹配形式的碱基片段。
以如下序列处理过程为例:
如步骤①所示,初始匹配片段长度为3,选取前三个碱基TAC作为匹配片段,由前的匹配方法可知对应四中片段为TAC,ATG,CAT,GTA。
通过往后检索,即可匹配到在右侧序列位置5、位置11、位置20出现了反向片段本身CTT,未出现其它对应形式片段。
3.2.可视化分析方法
在获得片段位置相关数据并进行统计分析后,得到了每组数据中片段的出现情况,并重点观察分析了出现次数最多的片段,为了进一步对序列中有对应形式的片段位置的确认以及进行观察,需要建立一套能够更为直观地对其中的精细特征进行观察的可视化模型。模型的结构图如图3所示。
输入:匹配模块输出的位置结果及选定需可视化的片段;
处理过程:在输入数据集片段位置结果以及选定片段后,首先需要将所有选定长度的片段位置结果提取出来,再从中选定指定的片段对应形式的位置,将其可视化。
而在匹配过程中,以X轴表示序列长度,Y轴表示不同序列,如数据集左右各N条序列,在第n组左右对应序列中,其长度为L,片段M在自匹配左侧序列中位置a,b(0
输出:针对自匹配所对应的片段位置图示以及自匹配和异匹配位置数据对应的片段对比位置图。
3.3.可视化结果及分析
在获得片段位置相关数据并进行统计分析后,得到了每组数据中片段的出现情况,并重点观察分析了出现次数最多的片段,如图4至图9所示。
通过上述可视化图示,可以观察出下列现象及特征:
(1)由数据集1中的图示可见,在数据集1中ATTTTT片段位置在序列中分布较为随机和均匀,而出现次数最多的AAGCTT以及其次的CCATGG在分布上具有十分明显的特征,它们在分布上自匹配和异匹配都有很明显的分界线,在序列大概50bp左右极少出现对应形式片段,大多出现在50~250bp区间段,之后成彗星尾部状出现次数及频率逐渐减少。
(2)从数据集2中的图示来看,呈现出了与数据集1中相似的特征与情况,AAGCTT与CCATGG分布十分相似,而TTTTTT则无明显现象。
(3)通过对数据集1和数据集2的对比中,两组数据呈现了相似的分布现象,出现次数最多的AAGCTT和CCATGG在另一组数据集中分布相似,说明了该特征不仅仅只局限于某一数据集。
(4)在对选取的其它数据集如部分编码区与非编码区的分析可视化中,并未发现较明显的类似现象与特征,这说明数据集1和数据集2所在的H2H基因可能具有一定的特殊性。
推荐阅读:基因多态性论文发表期刊推荐
4.结论
本文通过对DNA精细特征的分析处理,对DNA序列中出现的四种片段的结构进行了分析和整理,通过对专业处理后得到的数据集进行了匹配处理,得到了不同序列不同片段在数据集中的位置信息以及出现频率上的统计数据,在对这些结果进行分析后,确定了可视化的方法,选取了需要重点观察的片段对象,利用可视化方法对各数据集以及感兴趣的片段进行了自匹配可视化以及异匹配可视化展示。在最后的展示图示以及结果中,可以发现许多有意义的信息。从展示结果来看,部分片段如AAGCTT以及CCATGG在部分数据集中的分布以及数量可以为DNA序列的空间结构提供证据。同时更具体分布特性以及序列中的精细特征还可以进一步探索。
转载请注明来自:http://www.lunwencheng.com/lunwen/yix/16641.html
